
Comprendre les méthodes si on veut comprendre la portée d’une recherche ….
JTS aborde d’abord les méthodes – pour comprendre la portée des affirmations trouvées dans la vulgarisation : Comment on analyse l’ADN pour établir une généalogie ou un degré de filiation.
Ces techniques nouvelles révèlent des usages potentiels pour l’anthropologie, pour retrouver ses ancêtres, pour la police scientifique, et soulèvent des question éthiques – qui sont évoquées plus bas avec des publications développant ces problématiques
|
Des séquences spécifiques dans l’ADN mitochondrial sont très souvent utilisées. Comment on les lit et les compare été discuté dans une JTS précédente. ici
Et des données authentiques sont disponibles pour des activités en classe ici
On peut distinguer avec les élèves d’autres tests génétiques :les « empreintes génétiques » RFLP et STR
|
» Que ce soit un Serbe et un Suisse, ou un Finlandais et un Français, deux Européens ont probablement de nombreux ancêtres communs qui ont vécu il y a environ 1 000 ans. Une enquête génomique menée auprès de 2 257 personnes issues de 40 populations révèle que les personnes d’ascendance européenne sont plus étroitement liées les unes aux autres qu’on ne le pensait auparavant, et pourrait contribuer à apporter de nouvelles connaissances sur l’histoire européenne.
Les premiers efforts visant à retracer l’ascendance humaine grâce à l’ADN reposaient sur des « marqueurs génétiques uniparentaux » – des séquences d’ADN du génome mitochondrial, hérité de la mère, ou du chromosome Y, que les hommes héritent de leur père. »
« Ces études révèlent les grandes lignes de l’histoire humaine, comme la migration d’Homo sapiens hors d’Afrique il y a moins de 100 000 ans et sa colonisation ultérieure de l’Europe et de l’Asie. Mais les marqueurs uniparentaux ne contribuent pas à éclairer l’histoire plus récente, en partie parce qu’ils ne représentent qu’une seule lignée dans un arbre généalogique – comme la mère de la mère d’une mère, etc. « Callaway E., (2013) (traduit)
![]() encourage le lecteur à aller vérifier dans l’article d’origine : ici
encourage le lecteur à aller vérifier dans l’article d’origine : ici
Nous avons un ancêtre commun il n’y pas bien longtemps…
Un joli exemple pour des exercices de math ? « with high probability for large n, in each generation at least 1.77lgn generations before the present, all individuals who have any descendants among the present-day individuals are actually ancestors of all present-day individuals. (where lg denotes base-2 logarithm) » (Chang, J. T. (1999) Chang (1999) dit que selon le Wright–Fisher Model utilisés toutes les personnes qui vivaient à cette époque (et dont la descendance s’étend jusqu’à nous) sont en fait ancêtres de tous les n individus actuels de la population étudiée. Donc pour n ~ 9 millions, log2 (n) = 23,101 et 1.77 x log2 (n) = 40,889 (merci de me signaler une possible erreur) |
Comme on représente dans les arbres généalogiques tous les hommes portant le nom de famille et les femmes seulement en tant que descendantes d’un de ces hommes( c’est choquant, JTS partage votre indignation justifiée…), cette tradition cache le fait que nous avons un nombre énorme d’ancêtres (2 parents, 4 grands-parents, 8 arrière-grands-parents, etc.
Callaway, E. (2013 ici) l’explique ainsi « Sur la base de considérations théoriques, étant donné que chaque individu a 2n ancêtres d’il y a n générations, tous les humains seraient liés généalogiquement les uns aux autres sur des échelles de temps étonnamment courtes.
N.B: Ce Wright–Fisher Model suppose que les reproductions se font au hasard (panmixie) (Tataru et al., 2016 ici) et suppose qu’il n’y a pas de sélection, ni mutation, ni migration, et aucun mélange des générations, ce qui implique que père et mère appartiennent à la même génération.
Évidemment, ce modèle est une simplification du réel. Les chercheurs disposent maintenant de techniques permettant de dépasser ces limites: le séquençage qui permet de rechercher l’identité des fragments d’ADN issus de mêmes parents. .
Le séquençage permet une analyse bien plus fine
« Ces dernières années, les chercheurs se sont penchés sur le reste du génome – l’ADN qui peut provenir de l’un ou l’autre des parents – pour établir les « ancêtres génétiques ». C’est ce que Ralph et Coop (2013) ont fait pour reconstruire l’ascendance européenne . Leurs travaux sont publiés dans PLoS Biology ici. « We make use of genomic data for 2,257 Europeans (in the Population Reference Sample [POPRES] dataset) to conduct one of the first surveys of recent genealogical ancestry over the past 3,000 years at a continental scale. We detected 1.9 million shared long genomic segments, and used the lengths of these to infer the distribution of shared ancestors across time and geography. » ![]() encourage le lecteur à aller vérifier dans l’article d’origine : ici
encourage le lecteur à aller vérifier dans l’article d’origine : ici
« L’approche des chercheurs repose sur la manière dont les gènes sont remaniés à chaque génération, lorsqu’un individu forme de nouveaux ovules ou spermatozoïdes en mélangeant et en faisant correspondre les chromosomes hérités de chaque parent. » (Callaway, 2013) (traduit)
|
On se souvient que le crossing-over (Xover en abrégé) ou enjambement, résulte de l’échange – lors de la méiose (prophase I), d’un fragment d’ADN avec la chromatide homologue.
Cf. figure (Source : NIH). 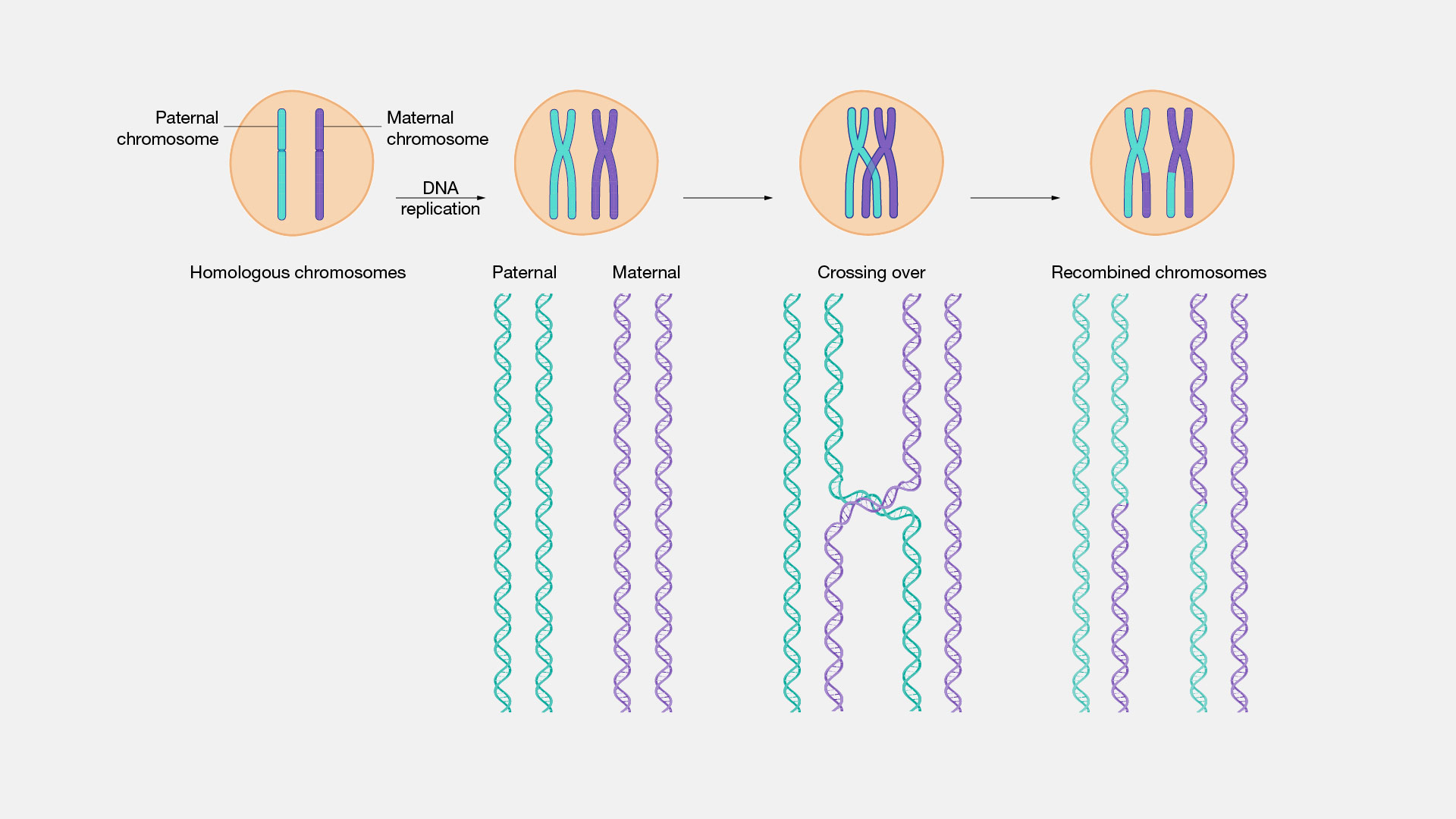 Ainsi des séquences d’ADN contiguës se trouvent séparées et ces fragments discontinus sont transmis indépendamment aux enfants.
Ainsi les segments transmis d’un seul tenant à la descendance sont réduits à chaque génération. La F1 n’a que quelques interruptions par rapport au parent. Plus on s’éloigne dans la descendance plus les fragments de cet individus ont été interrompus par le Xover.
Chaque chromosome subit entre un et trois Xover par méiose (source). |
» Les endroits où les séquences d’ADN sont échangées sont différents à chaque fois, de sorte que les segments ininterrompus transmis par une personne deviennent plus courts à chaque génération. Par exemple, les morceaux d’ADN partagés entre cousins germains sont plus longs que ceux partagés entre cousins germains, troisièmes et quatrièmes. » on shared long genomic segments, and used the lengths of these to infer the distribution of shared ancestors across time and geography. » ![]() encourage le lecteur à aller vérifier dans l’article d’origine : ici
encourage le lecteur à aller vérifier dans l’article d’origine : ici
Le séquençage permet de reconstituer des parentés en révélant les séquences d’ADN provenant d’un ancêtre commun
Si deux individus ont tous deux hérité d’une région génomique d’un ancêtre commun, cet ancêtre est appelé « ancêtre commun génétique » et la région génomique est partagée « à l’identique par descendance » (IBD) par ces deux individus. Nous définissons ici un « bloc IBD » comme étant un segment contigu du génome hérité (sur au moins un chromosome) d’un ancêtre commun partagé sans recombinaison intermédiaire (voir Figure 1A). […] Nous mesurons les longueurs des segments IBD en unités de Morgans (M) ou centiMorgans (cM), où 1 Morgan est défini comme étant la distance sur laquelle se produit une moyenne d’une recombinaison (c’est-à-dire un croisement) par méiose. » Ralph et Coop (2013) ici (traduit)
Pourquoi on ne se pose pas spontanément cette question ?En fait rares sont ceux qui se posent cette question… et Callaway (2013) ici ne développe guère comment on sait que tel fragment d’ADN est le même que tel autre.
Peut-être puisque nous ne différons que d’une infime partie de notre ADN (0.1% (Check Hayden, 2007) ici. Pourtant parait difficile de retracer un ADN avec de si infimes différences ? Voir aussi JTS ici pour clarifier les mesures de différence entre 2 humains ( Un inuit et moi différons de 0.1% mais je n’aurais que 50% de commun avec ma propre fille ??). Au contraire il parait naturel que les ADN soient différents, tant chacun des parents est différent. Cette conception est probablement renforcée par les schémas comme la Fig. 1 illustrant les fragments d’ADN par des couleur différentes. Cela renforce l’idée qu’un ADN issu d’untel est intrinsèquement différent de ceux d’un autre humain.
Giordan (1996) s’est beaucoup intéressé à la mise en évidence des ces conceptions et à leur ténacité. Elles sont un obstacle à l’apprentissage comme Bachelard (1934) l’avait montré il y a fort longtemps « Quand il se présente à la culture scientifique, l’esprit n’est jamais jeune. Il est même très vieux, car il a l’âge de ses préjugés » Pour aller plus loin Betz, et al. (2019) ici identifient 3 tendances spontanées de la psychologie humaine (« cognitive construals« ) à expliquer les phénomènes, le finalisme, l’anthropocentrisme, et l’essentialisme. Pour anticiper ou comprendre les difficultés d’apprentissage observées dans sa pratique enseignante (« chaque année les élèves font les mêmes erreurs !! » ), on peut s’appuyer sur ces catégories de conceptions naïves: dans ce cas l’essentialisme : « an unobservable essential property (an « underlying reality » or « true nature ») that conveys category identity« – à cause d’une nature profonde les objets, même les molécules conservent cette propriété essentielle des êtres qui les ont formés au cours de leurs transformations. Ainsi on peut s’attendre à ce que les élèves perçoivent les ADN d’origine différente comme intrinsèquement différents, sans ressentir le besoin de chercher quelles différences dans la séquence permettent de les distinguer.
|
Mais comment on reconnaît de qui est issu le génome, puisqu’ils sont quasiment identiques !!?
Pour identifier ces séquences IBD, « Sufficiently long segments of IBD can be identified as long, contiguous regions over which the two individuals are identical (or nearly identical) at a set of single nucleotide polymorphisms (SNPs) that segregate in the population. » ![]() encourage le lecteur à aller vérifier dans l’article d’origine : ici
encourage le lecteur à aller vérifier dans l’article d’origine : ici
- Pour visualiser ces SNP avec les élèves voir ce scénario du projet Bioinformatique : opportunités pour l’enseignement
Les chercheurs ont donc eu l’idée de séquencer pour chercher quels SNP on trouve le long de ces 1,9 millions de « shared long genomic segments ».
Cette méthode apporte de nouvelles données à l’histoire des migrations en Europe
« Des sociétés commerciales de séquençage génétique utilisent cette propriété pour connecter des cousins éloignés inscrits dans leurs bases de données généalogiques. Ralph et Coop (2013) ont recherché des parents encore plus éloignés en identifiant des parties du génome partagé par des personnes vivant dans toute l’Europe. En examinant la longueur de ces morceaux, les chercheurs ont pu déterminer approximativement quand vivait l’ancêtre commun des cousins éloignés.
Ils ont trouvé des ancêtres communs pour la plupart des populations il y a à peine 500 ans. Des séquences d’ADN plus anciennes reliaient cependant des Européens géographiquement plus éloignés.
Leurs travaux ont également révélé des signatures génétiques pour des événements clés de l’histoire européenne, tels que la migration des Huns vers l’Europe de l’Est au quatrième siècle et l’essor ultérieur des peuples de langue slave. Les habitants actuels des pays d’Europe de l’Est partagent de nombreux ancêtres qui vivaient il y a environ 1 500 ans. Les Italiens, quant à eux, sont liés aux autres populations européennes principalement par le biais d’individus ayant vécu il y a plus de 2 000 ans, peut-être en raison de l’isolement géographique du pays. » (Callaway, 2013) (traduit) ![]() encourage le lecteur à aller vérifier dans l’article d’origine : ici voir aussi Ralph & Coop (2013) ici
encourage le lecteur à aller vérifier dans l’article d’origine : ici voir aussi Ralph & Coop (2013) ici
D’autres exemples plus récents :
- L’arrestation du présumé « tueur de Golden State », un Californien accusé d’une série de viols et de meurtres vieux de plusieurs décennies. Pour le retrouver, les forces de l’ordre ont d’abord analysé avec la technique mentionnée plus haut – un échantillon d’ADN provenant de la scène du crime (du sang, des cheveux ou du sperme) pour rechercher des centaines de milliers de marqueurs ADN [les SNP ] qui varient selon les personnes, mais dont l’identité est dans de nombreux cas partagée avec des parents génétiques. Ils ont ensuite téléchargé ces données sur GEDmatch, une base de données en ligne gratuite où n’importe qui peut partager ses données génomiques provenant de sociétés de tests ADN grand public telles que 23andMe et Ancestry.com pour rechercher des proches qui auraient aussi soumis leur ADN.
Dans ce cas la recherche dans près d’un million de profils de GEDMatch a révélé plusieurs parents équivalent à des cousins au troisième degré du propriétaire d’ADN trouvé sur la scène du crime lié au tueur de Golden State. D’autres informations telles que les archives généalogiques, l’âge approximatif et les lieux du crime ont ensuite permis à la police de repérer une seule personne et finalement de l’arrêter. Adapté d’après Kaiser (2018), qui discute aussi les enjeux éthiques et les risques pour la vie privée encourage le lecteur à aller vérifier dans l’article d’origine : ici
encourage le lecteur à aller vérifier dans l’article d’origine : ici
Kaiser, J. (2018). We will find you : DNA search used to nab Golden State Killer can home in on about 60% of white Americans. Science. https://doi.org/10.1126/science.aav7021 - Cette technique ouvre de nouvelles possibilités en police scientifiques et soulève des question éthiques – qui sont discutées dans cet article de Science : « L’arrestation de Joseph James DeAngelo le 24 avril 2018 en tant que présumé tueur en série de l’État de Californie, soupçonné d’avoir commis plus d’une douzaine de meurtres et 50 viols, a soulevé de sérieuses questions sociétales relatives à la vie privée. La percée dans l’affaire est survenue lorsque les enquêteurs ont comparé l’ADN récupéré sur les victimes et les scènes de crime à d’autres profils ADN consultables dans une base de données généalogique gratuite appelée GEDmatch. Cela présente une situation différente de l’analyse de l’ADN des individus arrêtés ou condamnés pour certains crimes, qui est collectée dans le National DNA Index System (NDIS) des États-Unis à des fins judiciaires depuis 1989. La recherche dans une base de données non judiciaire à des fins d’application de la loi a attiré l’attention du public, de nombreux se demandant à quel point de telles recherches sont courantes, si elles sont légales, et ce que les consommateurs peuvent faire pour se protéger, ainsi que leurs familles, des regards inquisiteurs de la police. Les enquêteurs se précipitent déjà pour effectuer des recherches similaires sur GEDmatch dans d’autres affaires, ce qui rend impérative une enquête éthique et légale sur une telle utilisation. »Traduction automatique encourage le lecteur à aller vérifier dans l’article d’origine : ici
Ram, N., Guerrini, C. J., & McGuire, A. L. (2018). Genealogy databases and the future of criminal investigation. Science, 360(6393), 1078‑1079. https://doi.org/10.1126/science.aau1083 - Une vaste étude de l’ADN de centaines de sépultures en Hongrie retrace le plus long arbre généalogique connu et l’absence des filles. . Basé sur l’ADN, il s’étend sur neuf générations, c’est le plus long arbre généalogique jamais publié. Il révèle les coutumes reproductives de mystérieux cavaliers médiévaux. L’étude est le plus grand exemple d’une nouvelle tendance dans la recherche sur l’ADN ancien visant à étudier non seulement des individus isolés, mais aussi des communautés et des familles entières. Il comprend le plus long arbre généalogique basé sur l’ADN jamais publié, s’étendant sur neuf générations.Traduction automatique
Curry, A. (2024). Massive DNA study reveals mating customs of mysterious medieval horse riders.Science. https://doi.org/10.1126/science.z3n4jen
- Des anthropologues prennent les armes contre la « science des races » Lors de leur réunion annuelle, les anthropologues ont commencé à élaborer un manuel pour contrecarrer l’utilisation abusive et raciste de la recherche. Ils combattent l’idée erronée selon laquelle les humains sont divisés en quelques races distinctes. Ils soulignent que les gènes et les populations humaines présentent des schémas complexes de variation et de mélange.Traduction automatique
Price, M. (2024). Anthropologists take up arms against ‘race science’. Science. https://doi.org/10.1126/science.z6n7io6 - L’excellent et très récent ouvrage sur ce que ces tests définissent vraiment. Est-ce l’ethnicité – et toutes les dérives racistes, est-ce que l’ethnicité est fondée sur des bases biologiques ou sociales ? « In fact, what the tests are very good at doing is finding close relatives, and this is perhaps why the whole enterprise should be rebranded as family, not ancestry, testing. »
Kampourakis, K. (2023). Ancestry Reimagined : Dismantling the Myth of Genetic Ethnicities. Oxford University Press. ISBN : 978-0197656341
Remerciements
A Laura Weiss pour avoir vérifié les calculs et discuté pour clarifier plusieurs points.
Références:
- Bachelard, G. (1934). La formation de l’esprit scientifique. Vrin.
- Betz, N., Leffers, J. S., Thor, E. E. D., Fux, M., de Nesnera, K., Tanner, K. D., & Coley, J. D. (2019). Cognitive Construal-Consistent Instructor Language in the Undergraduate Biology Classroom. CBE—Life Sciences Education, 18(4), ar63. https://doi.org/10.1187/cbe.19-04-0076
- Callaway, E. (2013). Most Europeans share recent ancestors. Nature, nature.2013.12950. https://doi.org/10.1038/nature.2013.12950
- Chang, J. T. (1999). Recent common ancestors of all present-day individuals. Advances in Applied Probability, 31(4), 1002‑1026. https://doi.org/10.1239/aap/1029955256
- Check Hayden, Erika, (2007), So similar, yet so different. Nature News 17 October 2007 | Nature 449, 762-763 (2007) | https://doi.org/10.1038/449762a
-
Curry, A. (2024). Massive DNA study reveals mating customs of mysterious medieval horse riders.Science. https://doi.org/10.1126/science.z3n4jen
- Giordan, A. (1996). Les conceptions de l’apprenant. Sciences humaines Hors. Serie, 12, 6.
- Kaiser, J. (2018). We will find you : DNA search used to nab Golden State Killer can home in on about 60% of white Americans. Science. https://doi.org/10.1126/science.aav7021
- Price, M. (2024). Anthropologists take up arms against ‘race science’. Science. https://doi.org/10.1126/science.z6n7io6
- Ralph, P., & Coop, G. (2013). The Geography of Recent Genetic Ancestry across Europe. PLOS Biology, 11(5), e1001555. https://doi.org/10.1371/journal.pbio.1001555
- Ram, N., Guerrini, C. J., & McGuire, A. L. (2018). Genealogy databases and the future of criminal investigation. Science, 360(6393), 1078‑1079. https://doi.org/10.1126/science.aau1083
- Rohde, D. L. T., Olson, S., & Chang, J. T. (2004). Modelling the recent common ancestry of all living humans. Nature, 431(7008), 562‑566. https://doi.org/10.1038/nature02842
- Tataru, P., Simonsen, M., Bataillon, T., & Hobolth, A. (2016). Statistical Inference in the Wright–Fisher Model Using Allele Frequency Data. Systematic Biology, syw056. https://doi.org/10.1093/sysbio/syw056