
Quelques pistes pour exploiter des séquences d’ADN
Basé sur le séquençage d’ADN mitochondrial HVS1 lors de l’évènement Expériment@l Séquençage d’élèves AuthenTIC du 29 mars 2011. Rédigé par Marie-Claude Blatter ISB-SIB et F. Lombard, avec E. Poloni de l’AGP et P. Descombes .
De la bioinformatique en classe de biologie ? vous n’y pensez pas !
La bio-informatique fait partie du quotidien des chercheurs dans tous les chapitres de la biologie, et la manipulation d’Expériment@l non plus n’a guère de sens que pour permettre des analyses, des comparaisons de séquences. Des « expériences » in-silico en somme. Et la bonne nouvelle est qu’elles sont accessibles en classe avec un simple accès internet.
Que peut-on attendre de ces expériences ?
Un des objectifs est d’aider les élèves à faire le lien entre leur vie et les séquences , très abstraites. Le fait d’obtenir des séquences de vrais élèves pourrait les aider à établir des liens entre ce qui est enseigné et leur vécu personnel. Un autre est de permettre aux élèves de comparer eux-mêmes ces séquences pour voir les similitudes et les différences. Dans une perspective évolutive les similitudes pointent vers une origine commune et les différences indiquent une évolution indépendante. Explorer ces séquences peut aussi aider l’élève à se construire une représentation plus complète de l’ADN, du génome, des mécanismes de l’évolution.
…une séquence non-codante ?
Pour vérifier – ou au moins éprouver – le fait que cette séquence est non-codante on peut visualiser l’ADN mitochondrial et observer l’absence de gènes (indiqués en couleur) dans ce secteur du génome.
- Accéder au site Mapviewer (solution),
- Cliquer le symbole MT pour l’ADNmitochondrial, (solution), on voit tout le génome mitochondrial.
- Déplacer les petits curseurs (triangle blanc sur bleu
 ) pour zoomer sur la partie tout à droite où se trouve HVS1 entre 16060 et 16360 (solution).
) pour zoomer sur la partie tout à droite où se trouve HVS1 entre 16060 et 16360 (solution).
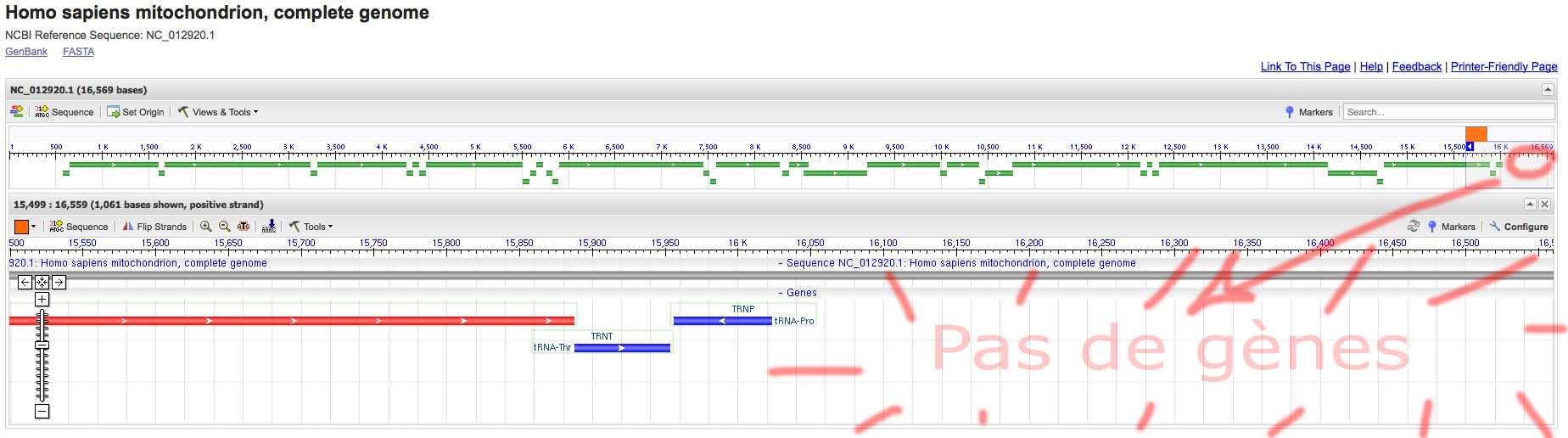 On observe que la séquence est dépourvue de gènes : non-codante
On observe que la séquence est dépourvue de gènes : non-codante
Aligner les séquences
- Se rendre sur le site Uniprot, Choisir l’onglet Align (Solution)
- Coller les séquences (elles sont en général au format Fasta) dans le cadre, cliquer le bouton Align ;
- La similitude des séquences apparait immédiatement. Un examen rapide montre des différences. L’étoile en bas de la liste révèle l’identité.
- Pour mieux voir les similitudes et les différences : cocher la case Similarity : on obtient un grisé plus foncé pour les zones les plus similaires(solution ici )
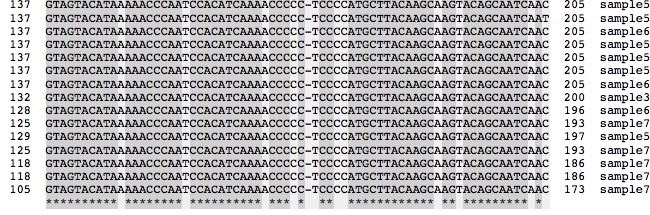 On peut discuter la signification des similitudes comme un indice d’origine commune et les différences comme signe de diversité génétique dans l’espèce.
On peut discuter la signification des similitudes comme un indice d’origine commune et les différences comme signe de diversité génétique dans l’espèce.
Le nombre de différences
Une analyse du nombre de différences par rapport à la séquence de référence rCRS donne déjà une mesure de la variabilité dans l’échantillon cf (ici): le nombre de mutations, en moyenne, entre deux séquences choisies au hasard peut être comparée aux moyennes de trois régions géographiques d’Europe (source : AGP ici):
- Sud-Est (3,8)
- Nord (3,5)
- Sud-Ouest (3,1)
L’alignement n’est pas une affaire simple
Si on veut travailler sérieusement avec ces séquences, il faut les aligner à la main : Ici on développera comment on a pu faire l’alignement. 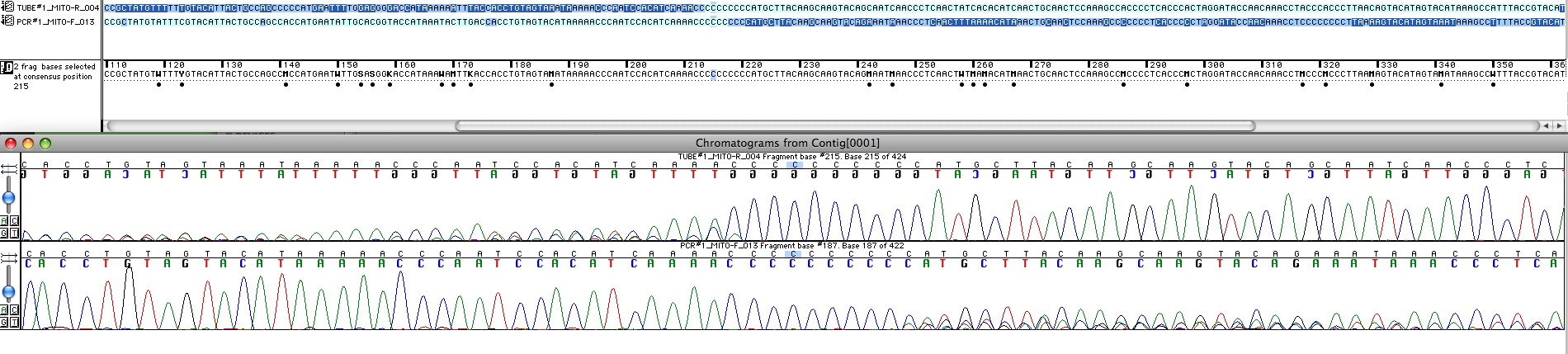 Fig x : L’alignement se fait à la main a partir du chromatogramme dans le logiciel d’alignement/edition. On voit une partie des séquences forward et reverse à leur jonction. En haut les séquences sont codées en bleu foncé pour les bases douteuses et bleu clair pour celles qui sont bien interprétables par le logiciel. En bas le chromatogramme. La séquence inférieure est bonne pour le début et celle de gauche pour la fin, la zone autour de 16189 reste douteuse.
Fig x : L’alignement se fait à la main a partir du chromatogramme dans le logiciel d’alignement/edition. On voit une partie des séquences forward et reverse à leur jonction. En haut les séquences sont codées en bleu foncé pour les bases douteuses et bleu clair pour celles qui sont bien interprétables par le logiciel. En bas le chromatogramme. La séquence inférieure est bonne pour le début et celle de gauche pour la fin, la zone autour de 16189 reste douteuse.
La glissade qui brouille le séquençage
Estella Poloni note que « pour 3 séquences ça a visiblement glissé vers 16180. » Cela signifie qu’une séquence avec une longue suite de C pose problème au séquençage Sanger qui « glisse » et la suite n’est pas fiable. De ce fait cette zone (environ 11 pb, des positions 16183 à 16193 inclues) est ignorée pour l’analyse du polymorphisme. Pour comprendre pourquoi, car c’est typiquement le genre de Q° qu’un élève malin peut poser, faisant preuve d’un esprit scientifique, qu’on ne voudrait pas étouffer, -> La glissade du séquençage qui rend flou
Un arbre de ces différences ?
Un tel arbre peut être obtenu à partir de l’alignement sur le site Phylogeny.fr : on peut s’inspirer du Scénario BIST 5 en l’adaptant. Cependant l’interprétation de cet arbre visuellement très prégnant, mais masquant aussi de nombreux choix – est une question complexe cf L’ADN mitochondrial, une petite molécule devenue star… E. Poloni dit qu’on risque « d’obtenir quelque chose de réticulé (un réseau plutôt qu’un arbre). En gros cela montre à quel point ça mute vite (ce qui fait que certains sites présentent des substitutions multiples ou récurrentes), et pour les élèves, ça peut être marrant de voir qui ressemble plus à qui… quant à en dire plus que ça… » Cela peut aider les élèves a mieux appréhender les études de génétique des population.
La même séquence existe-t-elle chez d’autre espèces?
A développer à partir du Scénario 5 en l’adaptant à cette séquence.
Comparer les protéines mitochondriales humaines actuelles et fossiles
- Toutes les protéines codées par la mitochondrie humaine (homo sapiens): @uniprot, solution
- Toutes les protéines codées par la mitochondrie néandertalienne (homo sapiens neandertal): @.uniprot.solution
- Toutes les protéines codées par la mitochondrie humaine Altaï (homo sapiens Altai): @uniprot.solution
Les amorces : vérifier leur spécificité
- Faire un Blast avec les amorces PCR pour vérifier que ces amorces « pêchent » bien la bonne séquence HVS1…
- Un gel a confirmé qu’avec ces amorces on amplifie bien un seul ADN dans chacun des 16 tubes :

Primer Forward L15996 5’-CTCCACCATTAGCACCCAAAGC-3’
Primer Reverse H16401 5’-TGATTTCACGGAGGATGGTG-3’
http://blast.ncbi.nlm.nih.gov/ choisir nucleotide blast: solution
Important: comme la séquence est courte, il faut choisir comme banque de données: Reference Genomic Sequences (RefSeq)
On obtient comme résultat pour CTCCACCATTAGCACCCAAAGC
NC_013993.1 Homo sp. Altai mitochondrion, complete genome (http://en.wikipedia.org/wiki/Denisova_hominin)
NC_011137.1 Homo sapiens neanderthalensis mitochondrion, complete genome NC_012920.1 Homo sapiens mitochondrion, complete genome
Les séquences stockées dans les banques de données sont pour la majorité 5′ -> 3’Pour l’autre amorce, il faut penser prendre la séquence inversée, soit: ACTAAAGTGC CTCCTACCAC
On obtient comme résultat: (le serveur du NCBI peut prendre du temps) -la séquence n’est pas spécifique à la mitochondrie !!!!, mais reconnaît aussi le chromosome X
| NW_002891894.1 |
Pongo abelii chromosome X genomic scaffold, P_pygmaeus_2.0.2
|
38.2 | 38.2 | 95% | 0.94 | 100% | |
| NT_167197.1 |
Homo sapiens chromosome X genomic contig, GRCh37.p2 reference primary assembly
|
38.2 | 94.6 | 95% | 0.94 | 100% | |
| NW_001218093.1 |
Macaca mulatta chromosome X genomic scaffold, Mmul_051212, whole genome shotgun sequence
|
38.2 | 38.2 | 95% | 0.94 | 100% | |
| NW_001842359.1 |
Homo sapiens chromosome X genomic contig, alternate assembly HuRef SCAF_1103279188171:1-1887445, whole genome shotgun sequence
|
38.2 | 38.2 | 95% | 0.94 | 100% | |
| NW_927700.1 |
Homo sapiens chromosome X genomic contig, alternate assembly Hs_Celera 211000035840903, whole genome shotgun sequence
|
38.2 | 94.6 | 95% | 0.94 | 100% | |
| NG_021256.1 |
Homo sapiens haloacid dehalogenase-like hydrolase domain containing 1 (HDHD1), RefSeqGene on chromosome X
|
38.2 | 38.2 | 95% | 0.94 | 100% | |
| NC_000023.10 |
Homo sapiens chromosome X, GRCh37.p2 primary reference assembly
|
38.2 | 207 | 100% | 0.94 | 100% | |
| AC_000155.1 |
Homo sapiens chromosome X, alternate assembly HuRef, whole genome shotgun sequence
|
38.2 | 179 | 100% | 0.94 | 100% | |
| AC_000066.1 |
Homo sapiens chromosome X, alternate assembly Hs_Celera, whole genome shotgun sequence
|
38.2 | 207 | 100% | 0.94 | 100% | |
| NC_012614.1 |
Pongo abelii chromosome X, P_pygmaeus_2.0.2
|
L’ADN mitochondrial contient plusieurs protéines
Toutes les protéines codées par la mitochondrie humaine (homo sapiens): @uniprot, solution
Ce qu’on a pu en faire
Le compte-rendu et l’analyse de l’activité faite par Bertrand Emery, maitre au Collège Calvin est ici : Au coeur de l’expérience, le récit d’une classe de 4e OC
Références
- Bendall, K. E., & Sykes, B. C. (1995). Length heteroplasmy in the first hypervariable segment of the human mtDNA control region. American journal of human genetics, 57(2), 248. pdf