
L’alignement n’est pas une affaire simple
Si on veut travailler sérieusement avec ces séquences, il faut ajuster les alignements à la main : Estella Poloni de l’AGP, a bien voulu partager avec Expériment@l ce travail d’alignement.
Le séquençage Sanger (complément ici : The Cell: A Molecular Approach. 2nd edition. Cooper GM.) donne des résultats bruts : la sortie du chromatogramme avec des pics de couleur pour chaque base. Cf. fig 1. Quand tout va bien, le logiciel attribue de manière fiable une base à chaque pic.
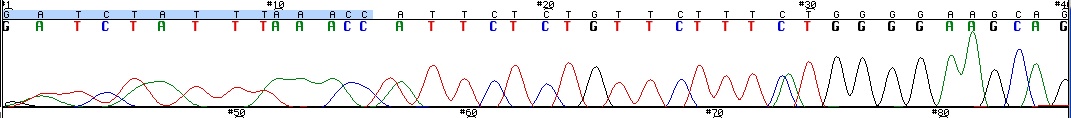
- Fig 1 : Chromatogramme d’un fragment. Le début de la séquence n’est pas utilisable, ce qui est normal avec la méthode Sanger.
De fait les données ne sont pas … données !
On voit bien que le début n’est pas clairement défini : on n’arrive pas à déterminer clairement une bonne douzaine de bases du début cf fig 1. La chercheure va donc éliminer les 14 bases du début.
Il est ainsi remarquable pour le non-expert de voir que la séquence n’est pas donnée : elle est une vérité scientifique, c’est-à-dire construite par une interprétation discutée et justifiée des données brutes.
L’automatisme fonctionne très bien dans la plupart des cas, l’oeil expert est nécessaire dans d’autres. Elle va donc revenir aux données brutes quand il faut choisir. Elle regarde la qualité du séquençage à partir du signal brut et elle choisit tantôt l’une tantôt l’autre des séquences : depuis un bout c’est la forward, et depuis l’autre bout c’est la reverse.
En cas de doute revenir au signal brut !
C’est un des fondements de la rigueur scientifique, la référence aux données brutes (ici le signal du chromatogramme) permet à l’expert de faire des choix éclairés et de produire des séquences qui sont sérieuses.
On voit dans la figure 2 qu’à partir de la position 235 environ du fragment, le signal n’est pas interprétable : des pics de plusieurs couleurs de même hauteur : même si le logiciel avait attribué des bases, mais elles ne sont pas signifiantes, la séquence n’est pas utilisable.
La signification de cette apparente non-unicité de l’ADN (Héteroplasmie) et des causes de cette difficulté est discutée ici -> La glissade du séquençage qui rend flou
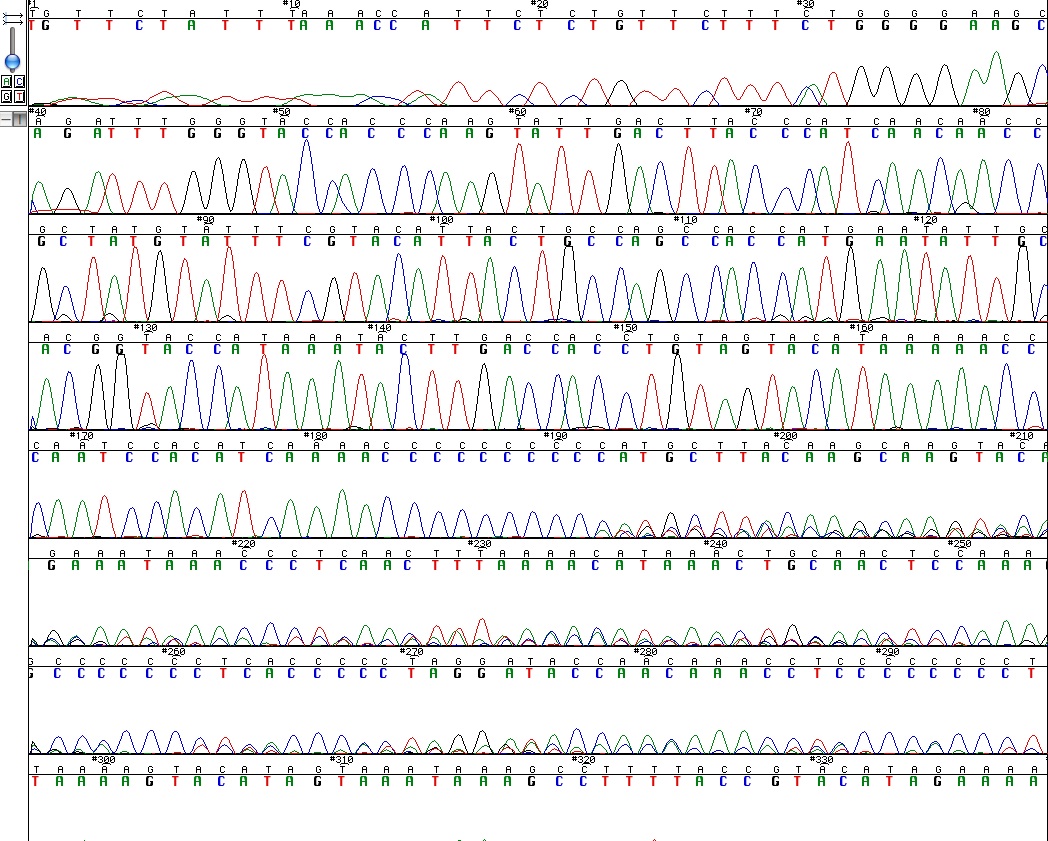
Fig 2 : Le chromatogramme d'une séquence. Le début est illisible et la fin inutilisable : c'est un échantillon où le séquençage a "glissé" : il a du être séquencé une deuxième fois.
Depuis chaque extrémité et rendez-vous au milieu !
Dans ces cas (3 dans notre échantillon), les ADN ont été séquencés à nouveau par C. Rossier dans l’autre sens, « reverse » en fait il s’agit d’un séquençage en avant, mais sur le brin complémentaire. On dispose alors de deux séquences qui contiennent de l’information fiable sur le début pour l’une et sur la fin pour l’autre. Il s’agit de les réunir autour de la zone incertaine vers 16189 qui reste toujours difficile.
On s’appuie alors sur la séquence de référence rCRS pour aligner : un logiciel (ici Sequencher) permet de la faire plus facilement en présentant les chromatogrammes, les bases synchronisées, et permet l’édition des bases.
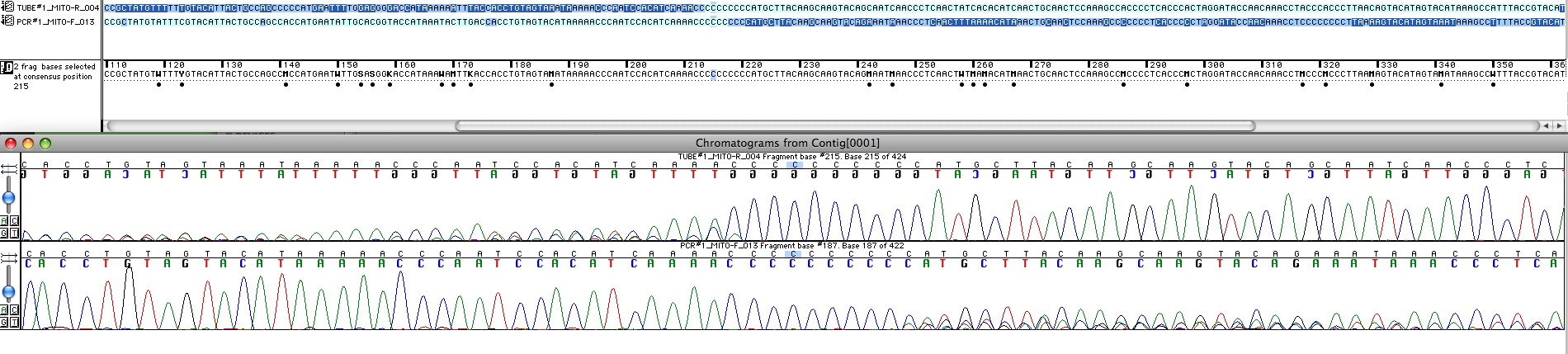
Fig 3 : L'alignement se fait à la main à partir du chromatogramme dans le logiciel d'alignement/édition. On voit une partie des séquences forward et reverse à leur jonction. En haut les séquences sont codées en bleu foncé pour les bases douteuses et bleu clair pour celles qui sont bien interprétables par le logiciel. En bas figure le chromatogramme correspondant. La séquence inférieure est bonne pour le début et celle de gauche pour la fin, la zone autour de 16189 reste douteuse.
Finalement on a un contig qui est le consensus des 2 mais où les séquences 16183 à 16193 sont ignorées. Ce travail a été effectué sur les 3 séquences glissées
L’alignement peut se faire une fois qu’on a des séquences propres.
Les 16 séquences de l’opération Expériment@l ont été mélangées aléatoirement à d’autres séquences de population genevoise pour obtenir un échantillon de 60 séquences anonymes. Elles sont disponibles ![]() ici.txt pour les membres S’inscrire à Expériment@l ?)
ici.txt pour les membres S’inscrire à Expériment@l ?)
Aligner les séquences demande de l’expertise
E. Poloni utilise un logiciel spécialisé onéreux mais Seaview est librement diffusé (Mac, Win, Linux). Ces logiciels alignent et permettent de mettre en évidence les différences.
Elle décide de ne conserver sur toutes les séquences que de 16057 à 16372 en éliminant le segment au milieu (env. 11 bases 16183 à 16193). Dans certains cas, elle a décalé en ajoutant ou en supprimant une base dans cette zone qu’on sait très susceptible de changements pour que le reste de la séquence s’aligne bien avec les autres. Il y a donc un choix de parsimonie ; on interprète qu’il y a une mutation dans cette zone et non quelques 150 mutations sur le reste de la séquence. Dans ce cas, on est assez sûr de cette interprétation, mais les données ne sont pas … données; elles sont construites.

Fig 4 : Alignement dans Seaview : les différences apparaissent en couleur
Comparer les séquences ?
Ces logiciels ont une fonction qui permet de mettre en évidence seulement les différences par rapport à la séquence de référence (cf. résultat ![]() ici pour les membres S’inscrire à Expériment@l ? ).
ici pour les membres S’inscrire à Expériment@l ? ).
Sans installer de logiciel, on peut exploiter des outils on-line pour mettre en évidence les changements : Uniprot en offre un très bon :
- Se rendre sur le site Uniprot, Choisir l’onglet Align (Solution)
- Obtenir les séquences disponibles
![[TXT]](http://tecfa.unige.ch/icons/text.gif) ici.txt pour les membres S’inscrire à Expériment@l ? ) au format Fasta
ici.txt pour les membres S’inscrire à Expériment@l ? ) au format Fasta - Coller les séquences dans le cadre, cliquer le bouton Align ;
- La similitude des séquences apparaît immédiatement. Un examen rapide montre des différences. L’étoile en bas de la liste révèle l’identité.
- Pour mieux voir les similitudes et les différences : cocher la case Similarity : on obtient un grisé plus foncé pour les zones les plus similaires (solution ici )

Marie-Claude Blatter du SIB-ISB note que UniProt est conçu normalement pour aligner des protéines (acides aminés), mais que ça marche quand même dans ce cas.
Les données ne sont pas données … elles sont construites
On voit bien dans ces exemples que les séquences produites ne sont pas données, elles sont construites et ne sont sérieuses que traitées avec sérieux par des gens compétents !
Références
- Gouy M., Guindon S. & Gascuel O. (2010) SeaView version 4 : a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular Biology and Evolution 27(2):221-224.