
Prédire la structure d’une protéine à partir de sa séquence,
puis déterminer la séquence d’une protéine qui aura la forme et la fonction désirée
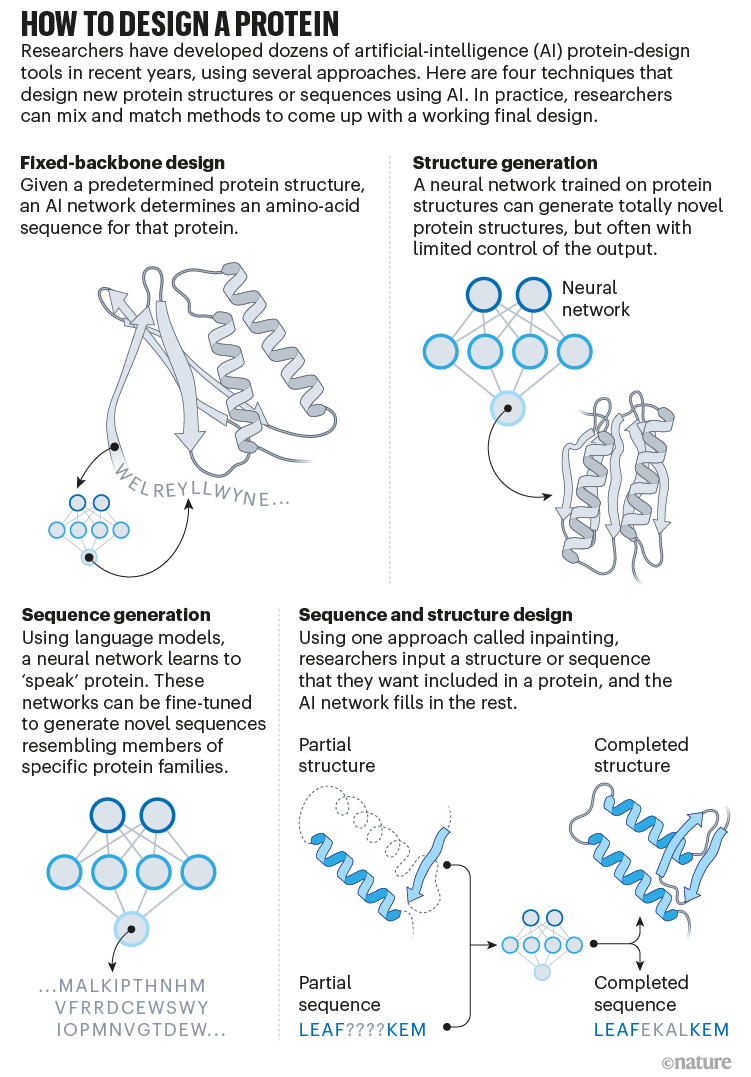
 Peu après cela l’équipe de Baker utilisant cette IA et d’autres ont pu faire l’inverse : trouver une séquence d’a.a. qui se repliera en prenant la forme d’une protéine souhaitée (médicament, vaccin, traitement de déchets etc.).
Peu après cela l’équipe de Baker utilisant cette IA et d’autres ont pu faire l’inverse : trouver une séquence d’a.a. qui se repliera en prenant la forme d’une protéine souhaitée (médicament, vaccin, traitement de déchets etc.).Ils ont même pu produire des protéines qui n’ont jamais existé par un procédé qu’ils ont joliment nommé « hallucination » (ils en font un verbe transitif : nous avons halluciné une protéine qui a telle fonction …). Leur procédé n’est pas sans ressembler à l’évolution : effectuer de nombreuses modifications aléatoires de la séquence ( comme les mutations et recombinaisons), puis prédire par IA la structure de chacune (comme l’expression du génome en phénotype) et choisir la séquence qui donner la structure la plus proche de ce qu’on cherche (comme la sélection naturelle), et recommencer jusqu’à satisfaction. Ensuite faire exprimer cette protéine dans une bactérie et vérifier son activité.
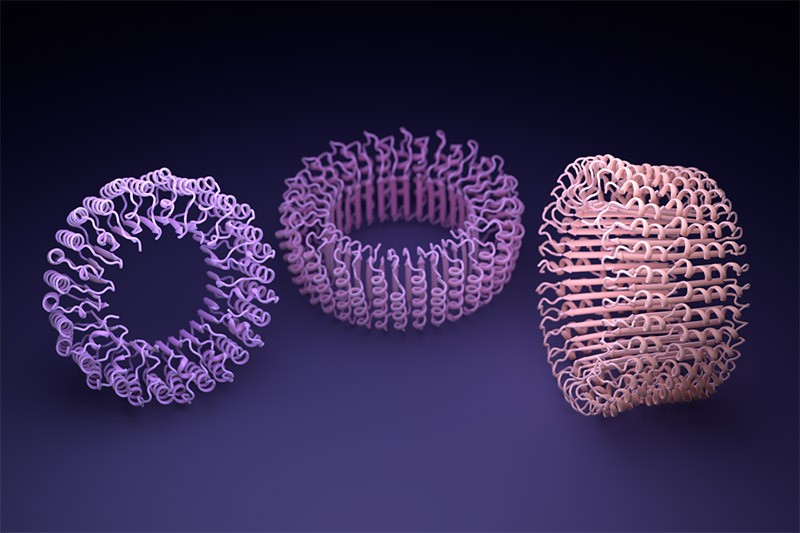
Ils sont récemment passé au niveau supérieur et ont même halluciné des assemblages de protéines (structure quaternaire) cf. fig 2 et ci-contre.
Implications pour l’enseignement de la biologie
JTS a sélectionné ces recherches parce qu’elles soulèvent plusieurs question importantes pour les enseignants. Elles sont importantes pour aider les élèves à comprendre ce que les médias en disent. Aussi parce que la biologie change, qu’elle est de plus en plus in silico mais s’articule avec la biologie moléculaire où ces prédictions reprennent contact avec le monde matériel pour vérifier leurs effets. Également parce que cela pose la question du naturel et des limites du vivant – qui définit en principe la biologie. Enfin parce que le métier de biologiste change pour devenir plus ingénieur ? C’est une différence de posture très significative.
Faut-il refléter ces changements dans nos cours ? Et comment ? JTS développe un peu ces questions plus bas.
« Nouvelle ère de la biologie numérique » : l’IA révèle les structures de presque toutes les protéines connues

Fig 1 : à droite Structure prédite par AlphaFold d’une protéine sur un stade d’un parasite du paludisme
Depuis, Hassabis, et al. (2022) ici ont dévoilé les structures probables de presque toutes les protéines connues – plus de 200 millions – des bactéries aux humains, une réalisation remarquable pour l’IA et un trésor potentiel pour le développement de médicaments et la recherche en biologie. (Travis, J. (2022), traduction Google par IA 😉 retouchée)
Un défi scientifique résolu ?
![]() encourage le lecteur à aller vérifier dans l’article d’origine :
encourage le lecteur à aller vérifier dans l’article d’origine :
- Tunyasuvunakool, K., Adler, J., Wu, Z., Green, T., Zielinski, M., Žídek, A., Bridgland, A., Cowie, A., Meyer, C., Laydon, A., Velankar, S., Kleywegt, G. J., Bateman, A., Evans, R., Pritzel, A., Figurnov, M., Ronneberger, O., Bates, R., Kohl, S. A. A., … Hassabis, D. (2021). Highly accurate protein structure prediction for the human proteome. Nature, 596(7873), 590‑596. https://doi.org/10.1038/s41586-021-03828-1
Un concert de superlatifs
« AlphaFold est une avancée capitale des sciences de la vie qui démontre la puissance de l’IA »
« Avec ce nouvel apport de structures qui révèle presque tout l’univers des protéines, nous pouvons nous attendre à ce que davantage de mystères biologiques soient résolus chaque jour. » (in Travis, J. (2022), traduction google translate (par IA 😉 retouchée)
Des potentiels fascinants …
Depuis une prédiction de ce qu’on pourrait observer …
vers une ingénierie de protéine originales composées à volonté
A peine remis de l’étonnement émerveillé de voir l’IA réussir à prédire la structure d’une protéine à partir de sa séquence, …(et peut-être perturbé par les implications dans nos cours ?) voici que des chercheurs ont réussi la fonction inverse : établir la séquence d’acides aminés qui donnera la forme d’une protéine souhaitée !
Fig 2: L’IA a permis aux chercheurs de produire des protéines qui ne ressemblent à rien de connu dans la nature [img].
Source : Callaway, E. (2022) credit Ian C Haydon/UW Institute for Protein Design
En quelques secondes plutôt que péniblement en plusieurs mois
(Callaway, E. (2022), dans une News de Nature écrit » le tout premier médicament à être fabriqué à partir d’une nouvelle protéine conçue par l’homme a été autorisé. Ce vaccin COVID 19 est basé sur une « nanoparticule » de protéine sphérique qui a été créée par des chercheurs il y a près de dix ans, grâce à un processus d’essais et d’erreurs très long et fastidieux. (Hsia, et al., 2016) ici.
![]() encourage le lecteur à aller vérifier dans l’article d’origine :
encourage le lecteur à aller vérifier dans l’article d’origine :
- Dauparas, J., Anishchenko, I., Bennett, N., Bai, H., Ragotte, R. J., Milles, L. F., Wicky, B. I. M., Courbet, A., de Haas, R. J., Bethel, N., Leung, P. J. Y., Huddy, T. F., Pellock, S., Tischer, D., Chan, F., Koepnick, B., Nguyen, H., Kang, A., Sankaran, B., … Baker, D. (2022). Robust deep learning–based protein sequence design using ProteinMPNN. Science, 0(0), eadd2187. https://doi.org/10.1126/science.add2187
- Wicky, B. I. M., Milles, L. F., Courbet, A., Ragotte, R. J., Dauparas, J., Kinfu, E., Tipps, S., Kibler, R. D., Baek, M., DiMaio, F., Li, X., Carter, L., Kang, A., Nguyen, H., Bera, A. K., & Baker, D. (2022). Hallucinating symmetric protein assemblies. Science, 0(0), eadd1964. https://doi.org/10.1126/science.add1964
Halluciner une protéine ! Halluciner devient un verbe transitif?
« De novo protein design by deep network hallucination »
JTS suggère qu’on peut le comparer aux mutations, recombinaisons, expression du génome en phénotype et à la sélection naturelle dans l’évolution.
 Fig 3: Production itérative de la protéine désirée. [img]. Source Baker Lab. (2021)
Fig 3: Production itérative de la protéine désirée. [img]. Source Baker Lab. (2021){kind=link}
Commencer par du charabia puis muter, prédire la forme, et sélectionner les plus adaptées.
Pour expliquer comment les réseaux de neurones « hallucinent » une nouvelle protéine, l’équipe de Baker la compare à la façon dont elle pourrait écrire un livre : « Vous commencez avec un assortiment aléatoire de mots – un charabia total. Ensuite, vous imposez une exigence telle que dans le paragraphe d’ouverture, il faut que ce soit une nuit sombre et orageuse. Ensuite, l’ordinateur changera les mots un par un et se demandera : « Est-ce que l’histoire a plus de sens ? ». Si c’est le cas, il conserve les modifications jusqu’à ce qu’une histoire complète soit écrite », Traduction de Baker Lab. (2022, juillet 21). Training A.I. to generate medicines and vaccines. Baker Lab.
- Anishchenko, … Baker, et al, (2021). De novo protein design by deep network hallucination. Nature, 600(7889), 547‑552. https://doi.org/10.1038/s41586-021-04184-w
Ils sont ensuite passé au wet lab la pour confirmer ces prédictions « nous avons caractérisé expérimentalement les hallucinations générées par ordinateur en obtenant des gènes synthétiques pour les 129 protéines, et en les exprimant et en les purifiant à partir d’E. coli »
Deux lignes de biologie moléculaire dans un article essentiellement de biologie numérique… La biologie change !
Designer une protéine c’est bien, en concevoir plusieurs s’assemblant, c’est mieux ?
- Wicky,…, Baker,et al., (2022). Hallucinating symmetric protein assemblies. Science, eadd1964. https://doi.org/10.1126/science.add1964
- Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., Wang, J., Cong, Q., Kinch, L. N., Schaeffer, R. D., Millán, C., Park, H., Adams, C., Glassman, C. R., DeGiovanni, A., Pereira, J. H., Rodrigues, A. V., van Dijk, A. A., Ebrecht, A. C., … Baker, D. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science, 373(6557), 871‑876. https://doi.org/10.1126/science.abj8754
Implications pour l’enseignement de la biologie
JTS estime que ces recherches soulèvent des question importantes et méritent l’attention des enseignants à plusieurs titres.
- Parce que c’est passionnant de voir une des grandes questions de la biologie obtenir une réponse… qui pose de nouvelles questions.
- Ces recherches sont reprises dans les médias, forcément simplifiées et en général surtout les conclusions sensationnalistes et les applications potentielles, mais pas les méthodes, le contexte et les limites. Les comprendre pour faire face à des questions d’élèves peut les aider à comprendre et … participer à leurs prises de décision en tant que citoyen-ne future ou actuelle.
- Parce que cela montre encore une fois que les savoirs de recherche que la recherche produit sont établis non plus par des opérations moléculaires, mais de biologie numérique (et peut-être apprivoiser le terme bioinformatique ?).
Si la biologie change profondément, la question de l’intégration dans nos programmes et pratiques en classe pourra-t-elle encore longtemps être écartée ?
- Parce que – quand même – à la fin on vérifie dans le « wet lab » par des techniques classiques de génie génétique si la protéine est bien celle attendue. In fine c’est dans le monde des molécules qu’on juge de la réussite. Hassabis (2022) dit que la mise à disposition de ces structures n’est qu’un point de départ. « Il y a encore évidemment beaucoup de biologie, et beaucoup de chimie, qui doivent être faites. » Ouf ! La biologie in silico – ou numérique ne remplace pas mais complète la biologie in vitro.
- Parce que la délimitation du naturel devient plus délicate : ces protéines synthétisée par des processus de biosynthèse classique – le génie génétique que nous avons découverts et non inventé peuvent encore être considérés comme naturels par certains, mais la production de protéines nouvelles n’existant pas dans la nature est-elle encore naturelle ? biologique ? fait-elle parie de l’étude du vivant ?
- Parce que la biologie devient avec la biologie de synthèse (cf. JTS Au-delà du génie génétique : la biologie synthétique. ) et ces travaux encore plus une ingénierie au service de production humaines – Callaway (2022) mentionne médicaments, vaccins, traitement des déchets.
- Parce qu’on s’éloigne encore plus du naturaliste contemplatif qui a dominé la biologie de la première moitié du siècle passé, de la biologie moléculaire, une recherche fondamentale, qui cherche à comprendre – en termes moléculaires – les mécanismes sous-jacents des phénomènes vivants. Avec cette nouvelle biologie, on ne se limite plus au vivant, on utilise la compréhension – ou du moins les possibilités d’application – pour produire de l’utile.
C’est un changement de posture très significatif. Faut-il le refléter dans nos cours ? Et comment ?
Un important chantier de réflexion sur l’essence de notre discipline mérite d’être ouvert – y participer avant qu’on nous impose une réforme ?
Références:
- Anishchenko, I., Pellock, S. J., Chidyausiku, T. M., Ramelot, T. A., Ovchinnikov, S., Hao, J., Bafna, K., Norn, C., Kang, A., Bera, A. K., DiMaio, F., Carter, L., Chow, C. M., Montelione, G. T., & Baker, D. (2021). De novo protein design by deep network hallucination. Nature, 600(7889), 547‑552. https://doi.org/10.1038/s41586-021-04184-w
- Baker Lab. (2021). Training A.I. to generate medicines and vaccines. Baker Lab (juillet 2021). https://www.bakerlab.org/2022/07/21/training-generate-medicines-vaccines/
-
Callaway, E. (2022). Scientists are using AI to dream up revolutionary new proteins. Nature. https://doi.org/10.1038/d41586-022-02947-7
- Dauparas, J., Anishchenko, I., Bennett, N., Bai, H., Ragotte, R. J., Milles, L. F., Wicky, B. I. M., Courbet, A., de Haas, R. J., Bethel, N., Leung, P. J. Y., Huddy, T. F., Pellock, S., Tischer, D., Chan, F., Koepnick, B., Nguyen, H., Kang, A., Sankaran, B., … Baker, D. (2022). Robust deep learning–based protein sequence design using ProteinMPNN. Science, 0(0), eadd2187. https://doi.org/10.1126/science.add2187
- Hassabis, D. (2022). AlphaFold reveals the structure of the protein universe. Consulté 5 août 2022, à l’adresse https://www.deepmind.com/blog/alphafold-reveals-the-structure-of-the-protein-universe
- Hsia, Y., Bale, J. B., Gonen, S., Shi, D., Sheffler, W., Fong, K. K., Nattermann, U., Xu, C., Huang, P.-S., Ravichandran, R., Yi, S., Davis, T. N., Gonen, T., King, N. P., & Baker, D. (2016). Design of a hyperstable 60-subunit protein icosahedron. Nature, 535(7610), 136‑139. https://doi.org/10.1038/nature18010
- Travis, J. (2022). ‘New era in digital biology’ : AI reveals structures of nearly all known proteins. Science. https://doi.org/10.1126/science.ade1829
- Tunyasuvunakool, K., Adler, J., Wu, Z., Green, T., Zielinski, M., Žídek, A., Bridgland, A., Cowie, A., Meyer, C., Laydon, A., Velankar, S., Kleywegt, G. J., Bateman, A., Evans, R., Pritzel, A., Figurnov, M., Ronneberger, O., Bates, R., Kohl, S. A. A., … Hassabis, D. (2021). Highly accurate protein structure prediction for the human proteome. Nature, 596(7873), 590‑596. https://doi.org/10.1038/s41586-021-03828-1
- Wicky, B. I. M., Milles, L. F., Courbet, A., Ragotte, R. J., Dauparas, J., Kinfu, E., Tipps, S., Kibler, R. D., Baek, M., DiMaio, F., Li, X., Carter, L., Kang, A., Nguyen, H., Bera, A. K., & Baker, D. (2022). Hallucinating symmetric protein assemblies. Science, 0(0), eadd1964. https://doi.org/10.1126/science.add1964