
Le consortium Telomere to Telomere a séquencé la partie du génome humain qui résistait encore au séquençage
Il y a 22 ans, une équipe de recherche internationale publiait séquence (quasi) complète du génome humain. Une véritable révolution qui permettait soudain d’avoir accès à l’ensemble – ou presque – de l’information génétique contenue dans nos cellules. On pouvait examiner une dimension cruciale de ce qui fait que nous sommes humains : LE génome humain.
Comme si nous avions tous le même génome ?
C’était tellement extraordinaire que rares étaient ceux qui posaient la question (cela paraissait presque gâter la fête) de la diversité génétique de l’humain. Cette question est devenue centrale et pour étudier cette différence une séquence de référence (GRCH38 actuellement) est nécessaire comme un étalon pour établir les différences (Cf. Fig 1).

En 2001 : Le génome humain … mais 8% d’information manquante
En 2001, le consortium scientifique international Human Genome Project était parvenu à séquencer la quasi-totalité de ce génome en identifiant trois milliards de bases. Cette découverte, révolutionnaire pour la médecine et la biologie, n’était cependant pas tout à fait complète. En raison de certaines limites technologiques, une petite partie – notamment les séquences répétitives – résistait en effet encore. On estimait alors que 5% à 15% de la séquence du génome humain restait à décoder.
Une séquence enfin complète ?
Le consortium Telomere to Telomere (T2T) auquel participe l’UNIGE est parvenu à séquencer les 8% manquants de notre matériel génétique (Nurk, et al., 2022) dans la revue Science ici. Le press release de l’UNIGE s’enthousiasme : « disposer d’une séquence complète, sans lacunes, des quelque 3 milliards de bases de notre ADN permet de comprendre le spectre complet des variations génomiques humaines et les contributions génétiques à certaines maladies. La génération d’une séquence du génome humain véritablement complète représente une incroyable réalisation scientifique, offrant la première vue complète de notre schéma d’ADN», déclare Eric Green, MD-PhD et directeur du NHGRI. «Cette information fondamentale renforcera les nombreux efforts en cours pour comprendre toutes les nuances fonctionnelles du génome humain, ce qui, à son tour, renforcera les études génétiques des maladies humaines.» Press release UNIGE
Sans réduire l’importance de cette étape dans notre connaissance du génome on relève qu’ils parlent d’un génome complet, alors que Nurk, et al. (2022) décrivent très factuellement cette (énorme il faut le reconnaitre) avancée mais aussi une limite de la séquence produite ici [la nouvelle séquence] « comprend des assemblages sans interruption pour tous les chromosomes sauf Y, corrige les erreurs dans les références antérieures, et introduit près de 200 millions de paires de bases de séquence… » Traduction d’un extrait de l’abstract.
On voit qu’un savoir scientifique définit ses limites et s’abstient de superlatifs – laissant le lecteur s’enthousiasmer – et JTS trouve qu’il y a de quoi.
T2T-CHM13 remplacera-t-il GRCH38 ?
La séquence de référence jusqu’à présent est nommée GRCH38. La comparaison avec la nouvelle séquence produite par T2T (CHM13) montre par exemple qu’elle inclut 3.5 milliards de paires de bases (Gbp) alors que GRCH38 en offrait 2.93 Cf Table 1 .![]() encourage le lecteur à aller vérifier dans l’article d’origine : ici
encourage le lecteur à aller vérifier dans l’article d’origine : ici
| STATISTICS | GRCH38 | T2T-CHM13 | DIFFERENCE (±%) |
|---|---|---|---|
| Summary | |||
| Assembled bases (Gbp) | 2.92 | 3.05 | +4.5 |
| Unplaced bases (Mbp) | 11.42 | 0 | −100.0 |
| Gap bases (Mbp) | 120.31 | 0 | −100.0 |
| Number of contigs | 949 | 24 | −97.5 |
| Contig NG50 (Mbp) | 56.41 | 154.26 | +173.5 |
| Number of issues | 230 | 46 | −80.0 |
| Issues (Mbp) | 230.43 | 8.18 | −96.5 |
| Gene annotation | |||
| Number of genes | 60,090 | 63,494 | +5.7 |
| Protein coding | 19,890 | 19,969 | +0.4 |
| Number of exclusive genes | 263 | 3,604 | |
| Protein coding | 63 | 140 | |
| Number of transcripts | 228,597 | 233,615 | +2.2 |
| Protein coding | 84,277 | 86,245 | +2.3 |
| Number of exclusive transcripts | 1,708 | 6,693 | |
| Protein coding | 829 | 2,780 | |
Table 1 source (Nurk,et al, 2022)ici
Le consortium T2T est parvenu à décoder les 8% manquants du génome humain complet (d’un télomère à l’autre : T2T) notamment grâce à des techniques nouvelles, voir plus bas un exemple avec Nanopore. Ce nouveau matériel génétique comprend plusieurs gènes mais aussi plusieurs séquences répétitives d’ADN.
En quoi les télomères sont-ils difficiles à séquencer ?
Ces séquences sont très répétitives. On peut le faire constater par les élèves dans une activité du projet Bioinformatique : opportunités pour l’enseignement où les élèves peuvent voir une authentique séquence de télomère dans le génome humain :
Pourquoi les séquences répétées sont-elles difficiles à séquencer ?
Le séquençage est souvent réalisé avec des appareils qui lisent des fragments assez courts, qui doivent ensuite être assemblés en alignant les chevauchements – c’est l’assemblage cf figure 2a.

La nécessité de cette opération n’est pas souvent comprise par les élèves et une activité permet de leur en faire comprendre la nécessité : exemple d’activité élève en figure 2b.
Une activité élève pour éprouver le mécanisme (l’algorithme) d’assemblage : choisir une séquence, la découper en bandelettes d’une douzaine de bases se chevauchant, et demander aux élèves de les assembler. On peut veiller à choisir des séquences avec peu de répétitions (séquence codantes en général) par exemple celle de l’insuline comme dans la fig b2.

Ensuite on peut le faire faire avec une séquence répétée pour comprendre la difficulté que T2T a affrontée. On voit alors l’immense difficulté et on constate que les séquences répétées étaient difficiles à assembler à partir de fragments courts. On voit aussi les possibilités qu’ouvrent les nouvelle technique (Nanopore voir plus bas) capables d séquencer de bien plus longues séquences
Le SIB propose parmi d’autres ici une activité « Médecine de précision et profil génétique » qui pourrait être détournée pour exercer l’alignement : A_vous_de_jouer.pdf Complément d’information ici
Ce qu’ont vraiment publié Nurk et al.

Le même numéro de Science publie plusieurs articles en rapport :
Pas seulement des répétitions, mais des gènes-clés identifiés dans les chromosomes acrocentriques
Nurk, et al. (2022) ont observé que la majeure partie de ces informations se situe près des télomères et des centromères, c’est à dire aux extrémités ou aux points de contact des chromatides. Il s’agit précisément des chromosomes 13, 14, 15, 21 et 22 acrocentriques (leur centromère se situe près d’une extrémité) et des chromosomes 1, 9, 16 et Y.
«Plus de la moitié des informations manquantes se situait sur les bras courts des chromosomes acrocentriques, qui contiennent les gènes de l’ADN ribosomal essentiels à la production de toutes nos protéines», explique Stylianos Antonarakis. Il précise qu’il s’agit «de gènes-clés pour la compréhension des mécanismes qui régissent le vieillissement et certaines maladies.»
Dans le cadre de T2T, Stylianos Antonarakis, de l’UNIGE, a été impliqué plus précisément dans le décodage d’un type spécifique de chromosomes dits «acrocentriques» dont il est spécialiste, et sur lesquels il a publié un review (Antonarakis, 2022) ici «Le premier papier date de 1934. Au total, une centaine d’études ont été passées en revue», indique-t-il. Avant la découverte du séquençage complet du génome, l’une des grandes questions que se posaient les scientifiques était notamment de savoir si les chromosomes acrocentriques étaient tous porteurs du même bras court en termes de matériel génétique.«La recherche du consortium T2T démontre aujourd’hui que le nombre de ces gènes est en réalité variable d’un être humain à l’autre», indique Stylianos Antonarakis. Ces découvertes ouvrent de nouvelles perspectives pour l’étude génétique de certaines maladies, de certains mécanismes liés au vieillissement et plus globalement des variations génomiques entre êtres humains.
Le séquençage Nanopore
Cette technologie est propriété de Oxford Nanopore Technologies et ce texte est tiré de leur site. Ils ont développé une technologie de séquençage d’ADN et d’ARN à base de nanopores protéiques, qui sont de minuscules trous canaux à travers les membranes. Dans leur technologie, les nanopores protéiques sont intégrés dans une membrane synthétique baignée dans une solution électrophysiologique et un courant ionique est passé à travers les nanopores.
Lorsque des molécules telles que l’ADN ou l’ARN se traversent ces nanopores, elles provoquent une perturbation du courant électrique. Ce signal peut être analysé en temps réel pour déterminer la séquence de bases des brins d’ADN ou d’ARN traversant le pore.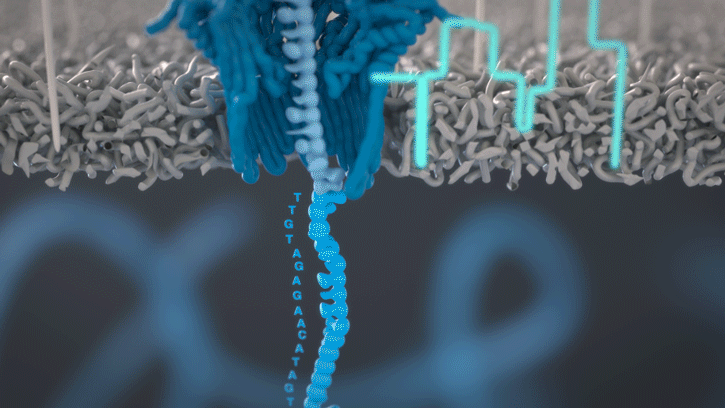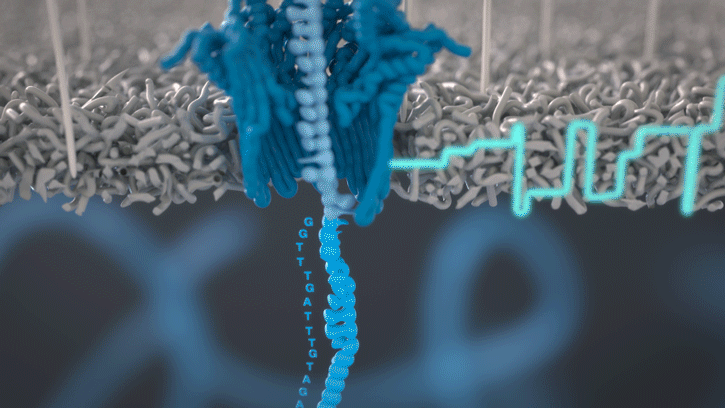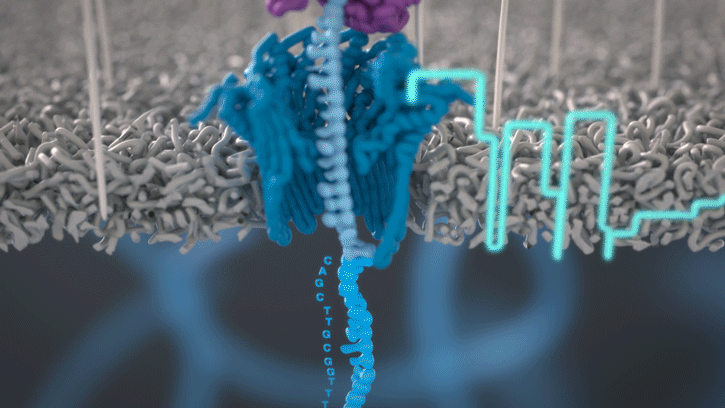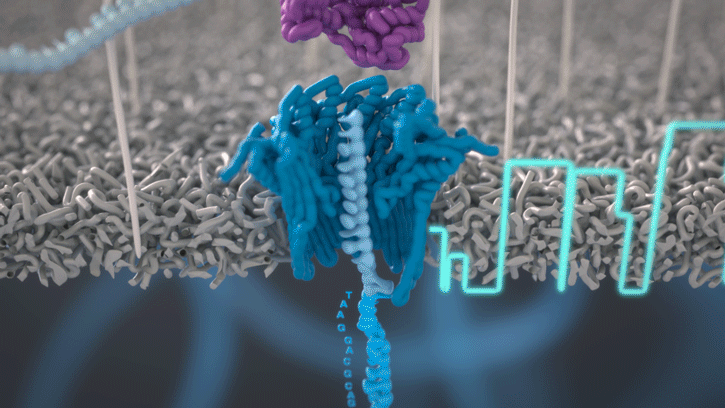
Fig 4: Séquençage d’un ARN : lorsque chaque base traverse le pore cela modifie le courant électrique à travers la membrane, ce qui permet de détermine quel nucléotide vient de passer. [img]. Source :Oxford nanopore
Sequençage de l’ADN et de l’ARN

Fig 6: Séquençage d’un ADN : un seul des brins traverse le pore. [img]. Source : Oxford nanopore
(Les membres Jump-To-Science peuvent obtenir ces articles…)
Références:
- Amarasinghe, S. L., Su, S., Dong, X., Zappia, L., Ritchie, M. E., & Gouil, Q. (2020). Opportunities and challenges in long-read sequencing data analysis. Genome Biology, 21(1), 30. https://doi.org/10.1186/s13059-020-1935-5
- Antonarakis, S. E. (2022). Short arms of human acrocentric chromosomes and the completion of the human genome sequence. Genome Research, 32(4), 599‑607. https://doi.org/10.1101/gr.275350.121
- Church, D. M. (2022). A next-generation human genome sequence. Science, 376(6588), 34‑35. https://doi.org/10.1126/science.abo5367
- Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., Vollger, M. R., Altemose, N., Uralsky, L., Gershman, A., Aganezov, S., Hoyt, S. J., Diekhans, M., Logsdon, G. A., Alonge, M., Antonarakis, S. E., Borchers, M., Bouffard, G. G., Brooks, S. Y., … Phillippy, A. M. (2022). The complete sequence of a human genome. Science, 376(6588), 44‑53.
Related Special Issue Research Articles - Oxford Nanopore Technologies, (2021) How nanopore sequencing works. (2021, novembre 29). Oxford Nanopore Technologies. http://nanoporetech.com/how-it-works
- Payne, A., Holmes, N., Rakyan, V., & Loose, M. (2018). Whale watching with BulkVis : A graphical viewer for Oxford Nanopore bulk fast5 files [Preprint]. Genomics. https://doi.org/10.1101/312256